| 1 |
听说酶
斯坦福大学医学院生物化学系的系主任Arthur Kornberg(阿瑟·科恩博格,1918-2007)一辈子研究酶,1959年获诺贝尔生理或医学奖。
1989年,他出版自传:《为了对酶的爱》(For the Love of Enzymes)--一位生物化学家的跋涉)

书中有一句话:从未见过一个乏味的酶(never a boring enzyme)。
这句话,对当时的神经生物学研究生我来说,非常陌生。但我记住了他这句话,虽然我当时非常不认同、不接受。
因为我的兴趣在神经系统,我们认为那才是令人激动的领域,而被迫背生化书里面的酶,那是要多乏味有多乏味。
我不仅认为神经生物学的科学问题明显比生物化学的问题更重要、更深远,而且当时我认为分子生物学和遗传学作为工具对神经生物学很重要。我要学分子生物学、遗传学,发现基因、发现基因编码的蛋白质参与神经的发育或功能。我的研究生论文就是这种思路的结果:克隆果蝇的一个基因big brain (bib),它参与脑的发育。
33年前,如果有人说,你以后不仅会认识到生物化学的重要性,而且会做纯粹是生物化学、不含神经生物学成分的研究,我会认为那人有精神病。
2022年,经过几年的研究,虽然最初出发点有神经生物学的原因,但我们发表了纯粹是生物化学的工作,而且是分离纯化的工作,居然是研究酶。
| 2 |
科恩博格老师
美国旧金山湾区,有三所大学在生物医学的世界前列:旧金山加州大学UCSF、伯克利加州大学UCB、斯坦福大学。它们之间交流较多。
UCSF因为全部是生物医学,所以如果有强项、也常常还有相当的体量。斯坦福因为有医学院,也有一定体量,但其医学院较小,研究的体量不是很大、而很精,临床的体量偏小。伯克利因为没有医学院,但其生命科学有些学科非常强。1980-1990年代,UCSF的老师用学生的选择来判断自己的竞争力。因为前几个学校基本竞争同一批学生,学生有效地用脚投票。当时的结果是UCSF、MIT和伯克利,其后是哈佛和斯坦福。这是UCSF老师告诉我们的、不可能没有偏见的数据。
斯坦福大学的生物化学系非常特别。它的创系系主任老科恩博格是纽约的犹太人。与那批犹二代一样,在贫穷的环境立志通过教育改善自己的命运。但他学了医以后却从医生改成做研究,二战期间曾经服役美国军舰,后到国立健康研究院(NIH)学了研究,他还到过圣路易斯的华盛顿大学,在两位捷克诺奖得主Carl Cori和Gerty Cori夫妇的实验室做实验。
1953年,科恩博格任华盛顿大学微生物系主任。那是华盛顿大学的生物化学的鼎盛时代,生物化学遍布多个基础和临床系科,重要发现层出不穷。就是神经生物学,也受生物化学明显影响。三位旅美犹太发育神经生物学家Victor Hamburger、Rita Levi-Montalcini、Stanley Cohen在华盛顿大学一系列工作,通过生物化学分离纯化到“神经生长因子”(NGF),是全世界第一个发现的生长因子。他们在研究过程中,得到了科恩伯格直接的建议和支持。
在华盛顿大学期间,科恩伯格课题组分离纯化了DNA多聚酶。1957年,他投稿《生物化学杂志》(JBC),被拒稿。哈佛大学的John Edsall听说后,告诉科恩博格:你别投其他刊物。我马上要成为JBC的主编了,我来发表你的文章。1958年,科恩博格实验室分离纯化DNA多聚酶的文章在JBC发表。1959年,科恩博格获诺贝尔奖。
同年,科恩博格到斯坦福大学医学院创立其生物化学系。他的博士后有个别成为系的教授主力。但他不是任人唯亲,而是为相对弱的斯坦福,争取到了非常好的教授。他带去的Paul Berg,因为对于分子生物学的贡献而获1980年诺奖。
斯坦福的生物化学系,很长时间在科恩伯格要求下,大家集体共同研究经费,这一奇葩做法,犹如以色列的kibbutz的美国科学版。当然,也是独此一家,而且后来不可能继续,但维持了很多年这一模式。
老科恩博格的三个儿子,两位是生物学家,一位是设计建造实验大楼的建筑师。他的大儿子Roger获2006年的诺贝尔化学奖,在老科恩博格去世的前一年。

二儿子Tom曾经被Julliard音乐学院录取,但后来也转而学生物。他父亲发现的细菌的第一个DNA多聚酶,被其他人发现不参与细菌常规的DNA复制。听上去是笑话。Tom发现细菌有三个DNA多聚酶,另外一个参与常规的DNA复制。看上去,他救了父亲。当然,这不过是生物化学常见的一个问题:有这样的作用,不等于在体内一定起最关键过程这样的作用。

我在UCSF读书期间,Tom是我们的“遗传学”课程的两位老师之一(另外一位是研究细菌和酵母的Ira Herskowitz,1946-2003)。我的果蝇遗传学理论部分是上他的课学的。他对果蝇发育基因的研究在世界上是最领先的几个课题组之一,特别是他和隔壁实验室合作首先证明所谓homeodomian是结合DNA而起调控果蝇发育的。这一基本概念一章到哺乳动物也是一样。
1980年代,我对遗传学和分子生物学是五体投地,认真学了。詹裕农、叶公杼实验室建立了很强的分子生物学和遗传学,我至少的认真努力地学了。
学了有多少效果,不能怪詹裕农、叶公杼老师,或Tom Kornberg老师,只能因为自己的智力限制。
而在整个研究生期间,对生物化学我都认为是老套过时。
| 3 |
对生化认识的渐变
对于生物化学的乏味认知,是逐渐改变的。
1994年,Marc Tessier-Lavigne在UCSF的实验室,通过生物化学分离纯化了吸引性分子netrin,推动神经纤维导向的分子机理的理解。(MTL后来曾任洛克菲勒大学校长、现任斯坦福大学校长)。
2004年,北京生命科学研究所成立前后,王晓东提议我们可以研究合作。我开始想哪些问题可以通过生物化学的分离纯化得以帮助、解决。我提的第一个课题是重编程:用蛙的卵细胞,从中分离纯化重编程因子,让成年分化的细胞核,可以再度成为全能干细胞。
王晓东雇了一位博士后,交代她在北生所准备这一实验。需要购买蛙,需要建立检测方法,等等。但这一课题后来没有下文。那时我们都没全职回国。
我到任北大几年之后,王晓东另有一位博士后(方敏)到北大任助理教授。几年后,因为照顾家庭方敏回美国了。据说,方敏离开时,最好的学生给了王晓东。次好的三位放我实验室。其中一位很快去美国,另一位也很快离开实验室,剩下刘玉祥。用方敏的课题获得博士学位后,他留在我实验室。
| 4 |
生化研究的实践
我长年讲生物钟的课,因为生物钟的分子机理有趣和重要。也因为我专门生物钟的研究用来讲遗传筛选对于生物学研究的重要性,现在是《生物学概念与途径》的第14章。讲课久了,总结研究会发现,其中的生物化学方法用的很少,但看起来可以用了。人的Per2基因突变,导致人的生物钟改变。点突变改变的是磷酸化位点,特别是S662。我先建议研究生余腾辉筛选磷酸化S662的蛋白激酶。他和我都不会生物化学,我们用分子生物学的方法,把编码每一个蛋白激酶的基因(cDNA),都轮流与Per2共转染,看看哪一个激酶可以磷酸化S662。但那时,我们试了好几次,拿不到针对磷酸化S662的抗体,所以不能检测S662是否磷酸化了(pS662)。余腾辉最后看了另外一个位点,发现了一个可以磷酸化这个位点的蛋白激酶。
我们听王晓东说,北生所与余国良的公司有合约,可以不费钱而由后者的公司给打抗体,如果能够说服他们这种抗体有销路。建议者不用付费,其他人要付费。我们后来两次通过这一途径,每一次都很顺利得到针对特定蛋白质、特定磷酸化位点的抗体。
这样,我们也有了对pS662的抗体。刘玉祥和研究生李扬再次用分子生物学的方法筛选,确定了一个磷酸化S662的蛋白激酶。因为这个位点的蛋白激酶,有19年都没有人发表文章找到,所以我们得意洋洋,不紧不慢,写了文章也不投稿,而还想做的更好。结果,一个外国实验室在PNAS发表文章,找到同一蛋白激酶。结果不如我们的全、不如我们的漂亮,但结论是一样的。我们这篇文章,除了给一两个朋友看,就没有投稿,还在我计算机上。
日本筑波的科学家2016年发现SIK3参与睡眠。而SIK3本身有几个磷酸化位点。日本科学家集中研究556位点。刘玉祥、李扬集中研究T221位点。我们用抗体识别pT221。李扬集中研究T221磷酸化及其生物学功能(睡眠)。
刘玉祥带领王涛等找可以磷酸化SIK3-T221的蛋白激酶。SIK3是AMPK家族成员。AMPK在代谢中其非同寻常的重要作用。2003年,几个英国和美国实验室发现LKB1可以磷酸化AMPK-T172,其后发现也可以磷酸化SIK3-T221。
研究生刘子怡研究了果蝇和小鼠的LKB1,发现它促进果蝇和小鼠的睡眠(文章将见于美国遗传学会的《遗传学》杂志)。
刘玉祥、王涛等发现除外LKB1,还有可以磷酸化SIK3-T221的活性。他们决定用蛋白质分离纯化的途径,找SIK3-T221的蛋白激酶。通过经典的生物化学方法,他们找到了MST3。
这一发现,是经典的生物化学的成功。
| 5 |
回望与前路
我实验室长期用遗传学方法研究神经生物学。优点的能够确定功能,缺点是不一定理解分子机理。例如,我的研究生论文,发现参与果蝇脑发育的big brain基因,如果遗传突变导致果蝇缺失这一基因,果蝇的脑就特别大。这一基因的功能很重要。但有了基因功能,还需要确定其机理。拿到bib基因后,分析序列,当时以为是甘油转运蛋白,后来知道它编码水通道蛋白。但是,水通道蛋白如何调控神经发育,机理连不上,因此迄今理解有限。
而用生物化学的优点是,理解分子在分子层面是做什么的,但不一定理解它在生物学上的功能及意义。例如,发现MST可以磷酸化SIK3-T221和AMPK-T172,是生物化学的发现。但不清楚哪个MST在细胞内确实起磷酸化SIK3-T221和AMPK-T172的作用。也不清楚如果有这样的磷酸化反应,可以调控什么生物学功能(能够调节睡眠吗?能够调节代谢吗)。需要更多后续的研究。
有时候,可以疑问自己到底是成熟了,还是老糊涂了。
科学道路上,认知有改变,完全不用引以为耻,而不排除引以为荣的可能性。




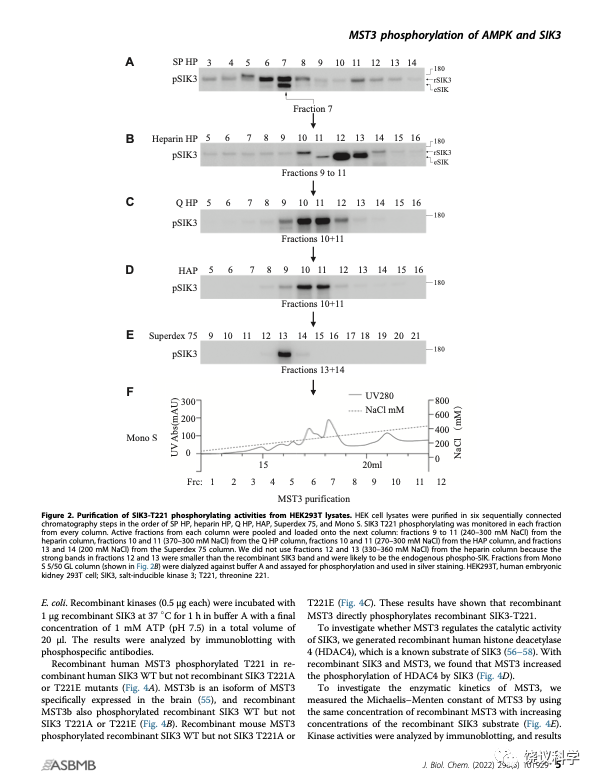
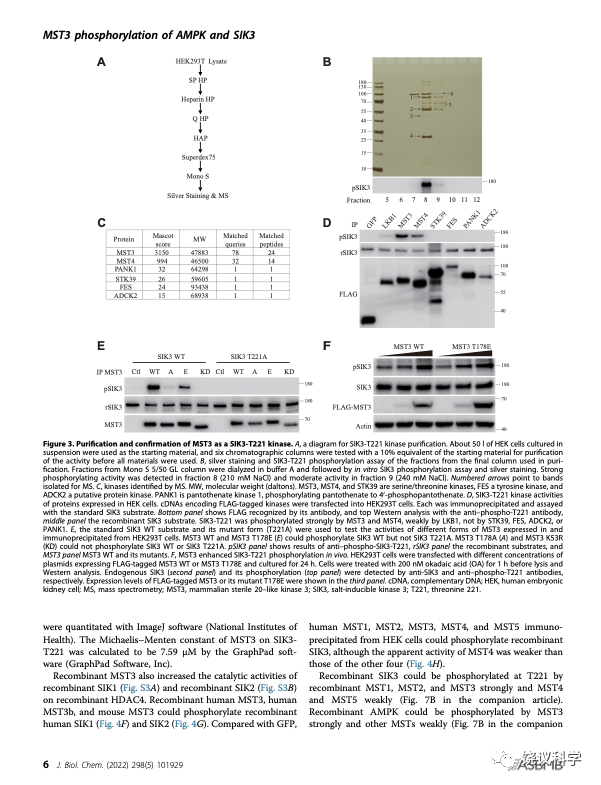
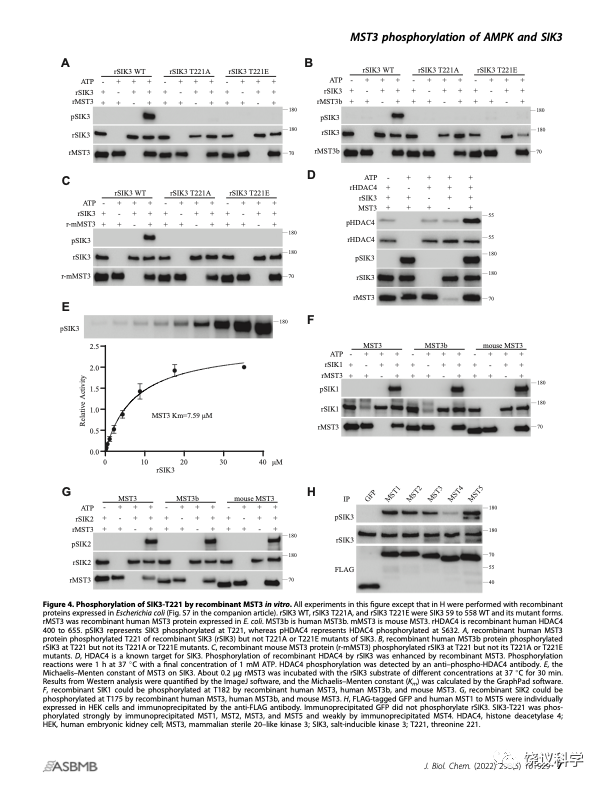

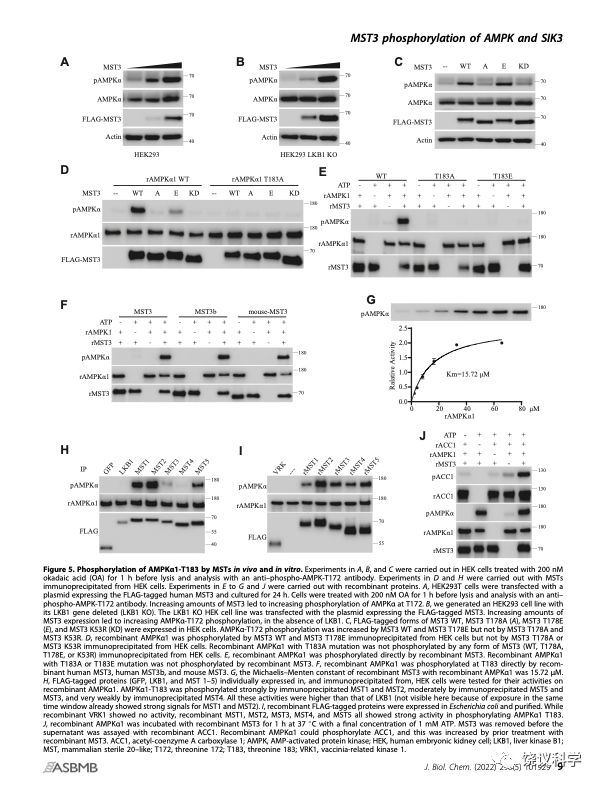



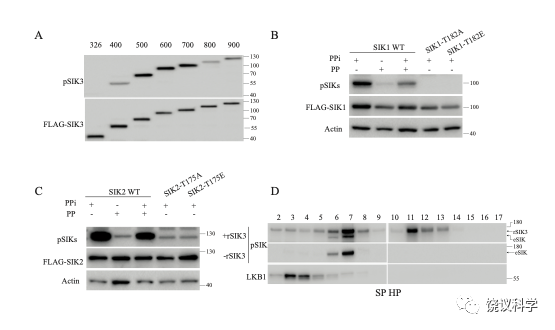
Figure S1. SIK substrates for phosphorylation and rSIK3 vs eSIK in HEK cells. A, T221 phosphorylation of truncated SIK3 fragments. HEK293T cells were transfected with plasmids encoding SIK3 fragments of different lengths (indicated by the numbers) (the exact aa begins with aa 59, ending with 385, 458, 558, 658, 758, 858 and 958, respectively). Cells were harvested 24 h after transfection and extracts were heated at 95 ℃ with the protein loading buffer and analyzed with the monoclonal anti-phospho-T221 antibody (mAb). Fragment 59 to 558 of SIK3 was chosen and generated from E. coli as the standard substrate. B, The mAb recognizing phospho-T221 of SIK3 also recognized phospho-T182 of SIK1. HEK293T cells were transfected with SIK1 plasmids and harvested 24 h after transfection and analyzed by Western analysis with the mAb. PP indicated λ-protein phosphatase and PPi indicated phosphatase inhibitor. C, The mAb recognizing phospho-T221 of SIK3 also recognized phospho-T175 of SIK2. HEK293T cells were transfected with SIK1 plasmids and harvested 24 h after transfection and analyzed by Western analysis with the mAb. D, SIK3 T221 phosphorylating activities in HEK cells. HEK cell lysates with 500 mg proteins (in a concentration of 10 mg/ml) were filtered at 0.45 mm and fractionated through a cationic SP HP column. Fractions were collected and aliquots of each fraction were dialyzed against buffer A, followed by in vitro SIK3 phosphorylation assay. Recombinant SIK3 was added to the experiments shown in the upper panel, which shows two bands in pSIK (rSIK and eSIK). Recombinant SIK3 was not added to the middle panel, which shows only one band, an endogenous SIK band (eSIK).
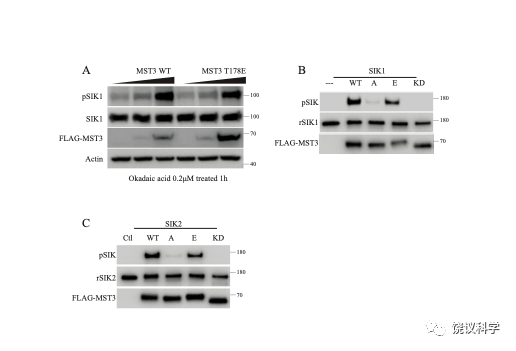
Figure S2. Phosphorylation of SIK1 and SIK2 by MST3. A, MST3 enhanced SIK1-T182 phosphorylation in vivo. HEK293T cells were transfected with different concentrations of plasmids expressing MST3 WT or T178E and cultured for 24 h. Cells were treated with 200 nM OA for 1 h before lysis and analysis of SIK1-T182 phosphorylation. Endogenous SIK2 was too low to be detectable in this experiment. B, Recombinant SIK1 was phosphorylated by MST3 WT and MST3 T178E immunoprecipitated from HEK cells, but not by MST3 T178A or K53R immunoprecipitated from HEK cells. C, Recombinant SIK2 was phosphorylated by MST3 WT and MST3 T178E immunoprecipitated from HEK cells, but not by MST3 T178A or K53R immunoprecipitated from HEK cells.
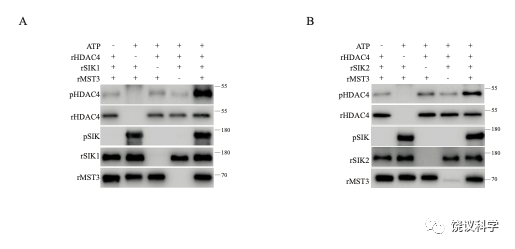
Figure S3. Catalytic activities of SIK1 and SIK2 increased by recombinant MST3. A recombinant SIK was made in E. coli as a fusion protein in the form of MBP-TEV-3xFLAG tag-SIK-TEV-GFP-8xHis tag. Before being used in experiments here, the recombinant fusion protein was cleaved at the two TEV sites and thus SIK was released without the fusion proteins or the tags. A, Recombinant SIK1, recombinant MST3, and recombinant HDAC4 were incubated for 1 h at 37 ℃ with a final concentration of 1 mM ATP. HDAC4 phosphorylation was detected by an antibody against phospho-HDAC4. The presence of recombinant MST3 increased the activity of recombinant SIK1 on recombinant HDAC. B,Recombinant SIK2, recombinant MST3, and recombinant HDAC4 were incubated for 1 h at 37 ℃ with a final concentration of 1 mM ATP. HDAC4 phosphorylation was detected by an antibody against phospho-HDAC4. The presence of recombinant MST3 increased the activity of recombinant SIK2 on recombinant HDAC.

Figure S4. Phosphorylation of AMPKα2-T172 by MST3 in vivo and in vitro. A, MST3 KO A7 is an HEK cell line with its MST3 gene deleted by gene targeting. MST3 enhanced the phosphorylation of AMPKα-T172 in HEK cells. Increasing concentrations of the plasmid expressing MST3 WT were transfected into these MST3 KO cells and cultured for 24 h. Cells were treated with 200 nM OA for 1 h before lysis and analysis with the anti-phospho-AMPKα-T172 antibody. B, MST3 KO B4 is another HEK cell line with its MST3 gene deleted, similar to Fig. S4A. C, Phosphorylation of AMPKα1 is required for its catalytic activity. FLAG-tagged AMPKα1 was immunoprecipitated from HEK cells and pre-treated with or without λ-phosphatase for 1 h at 37℃. λ-phosphatase was removed before the supernatant was tested for ACC1 phosphorylation which was detected by an antibody against phospho-ACC1. D, The catalytic activity of AMPKα1 WT on ACC1 was higher than those of T183E and T183A mutants of AMPKα1 immunoprecipitated from HEK cells after transfection. E, AMPKα1 enzymatic activity requires T183 phosphorylation. Recombinant AMPKα1 WT, T183A, T183E proteins expressed by and purified from E. coli were assayed with recombinant ACC1 for 1 h at 37 ℃ with a final concentration of 1 mM ATP. ACC1 phosphorylation was detected by an antibody against phospho-ACC1. F,Immunoprecipitated MST3 enhanced the phosphorylation of AMPKα2 at T172 in vitro. MST3 WT and MST3 mutants T178A, T178E, K53R were immunoprecipitated from HEK293T and assayed with recombinant AMPKα2. G,Recombinant MST3 directly phosphorylated recombinant AMPKα2 WT at T172 but not T172A and T172E mutants of AMPKα2. H, Recombinant human MST3b and recombinant mouse MST3 directly phosphorylated AMPKα2-T172 in vitro. (I) The Michaelis-Menten constant of recombinant MST3 on recombinant AMPKα2-T172 was determined to be 19.87 uM.
